
Published in: Journal of Medicinal Chemistry, ASAP
DOI: 10.1021/acs.jmedchem.4c01241
Authors: Trent D. Ashton, Petar P. S. Calic, Madeline G. Dans, Zi Kang Ooi, Qingmiao Zhou, Josephine Palandri, Katie Loi, Kate E. Jarman, Deyun Qiu, Adele M. Lehane, Bikash Chandra Maity[TCGLS Member], Nirupam De[TCGLS Member],Carlo Giannangelo, Christopher A. MacRaild, Darren J. Creek, Emma Y. Mao, Maria R. Gancheva, Danny W. Wilson, Mrittika Chowdury, Tania F. de Koning-Ward, Mufuliat T. Famodimu,Michael J. Delves, Harry Pollard, Colin J. Sutherland, Delphine Baud, Stephen Brand, Paul F. Jackson, Alan F. Cowman, and Brad E. Sleebs
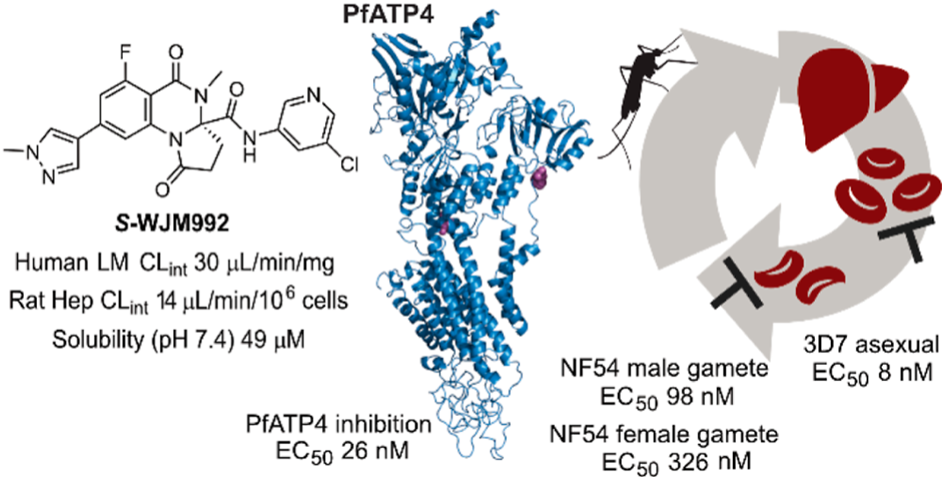
Abstract: To contribute to the global effort to develop new antimalarial therapies, we previously disclosed initial findings on the optimization of the dihydroquinazolinone-3-carboxamide class that targets PfATP4. Here we report on refining the aqueous solubility and metabolic stability to improve the pharmacokinetic profile and consequently in vivo efficacy. We show that the incorporation of heterocycle systems in the 8-position of the scaffold was found to provide the greatest attainable balance between parasite activity, aqueous solubility, and metabolic stability. Optimized analogs, including the frontrunner compound S-WJM992, were shown to inhibit PfATP4-associated Na+-ATPase activity, gave rise to a metabolic signature consistent with PfATP4 inhibition, and displayed altered activities against parasites with mutations in PfATP4. Finally, S-WJM992 showed appreciableefficacy in a malaria mouse model and blocked gamete development preventing transmission to mosquitoes. Importantly, further optimization of the dihydroquinazolinone class is required to deliver a candidate with improved pharmacokinetic and risk of resistance profiles.
Graphical Abstract


@ 2021 TCG Lifesciences Private Limited. All Right Reserved.
.jpg ?>)
.jpg ?>)
