
Published in: J. Med. Chem. 2025, 68, 21, 23485–23520
DOI: 10.1021/acs.jmedchem.5c02394
Authors: Suraksha Gahalawat, Sneha Ray, Xiaoyu Zhang, Xiaoyi Deng, Yan Han, Zhe Chen, Aloysus Lawong, David M. Shackleford, Kasiram Katneni, Gong Chen, Peng Li, Alice Ng, Longjin Zhong, Meiyu Hu, Mitchell McInerney, Wen Wang, Jessica Saunders, Daniel Collins, Jaya Jayaseelan, Cassandra L. Noack, Bikash C Maity [TCGLS Member], Nirupam De [TCGLS Member], Benoît Laleu, Simon F. Campbell, Margaret A. Phillips,* Susan A. Charman,* and Joseph M. Ready
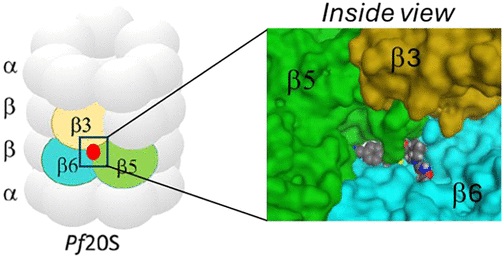
Abstract: Malaria remains a critical global health challenge, with increasing resistance to frontline therapies necessitating novel drug targets. The proteasome has emerged as a promising target for antimalarial drug discovery. This study describes efforts to optimize a series of species-selective reversible inhibitors targeting the Plasmodium falciparum 20S proteasome. Starting from the carboxypiperidine scaffold identified through a high-throughput viability screen, we conducted iterative structure−activity relationship studies, leading to the development of highly potent and selective inhibitors with good oral bioavailability. Lead compounds demonstrated nanomolar potency against P. falciparum blood-stage parasites and selective inhibition of the parasite proteasome over the human counterpart. Cryo-EM structural studies confirmed binding at the β5 subunit, while in vivo pharmacokinetic studies identified promising candidates for further development. These findings support proteasome inhibition as a viable strategy for novel antimalarial drug development.


@ 2021 TCG Lifesciences Private Limited. All Right Reserved.
.jpg ?>)
.jpg ?>)
